- Integrated, intuitive, property oriented GUI for building molecules and larger systems:
- Automated 2D -> 3D conversion with hydrogen addition.
- Library of popular organic molecules.
- Protein editor / peptide builder.
- Crystal builder.
- Builders for Molecular Dynamics systems (Solid state, liquid, amorphous, polymer, liquid crystal and more).
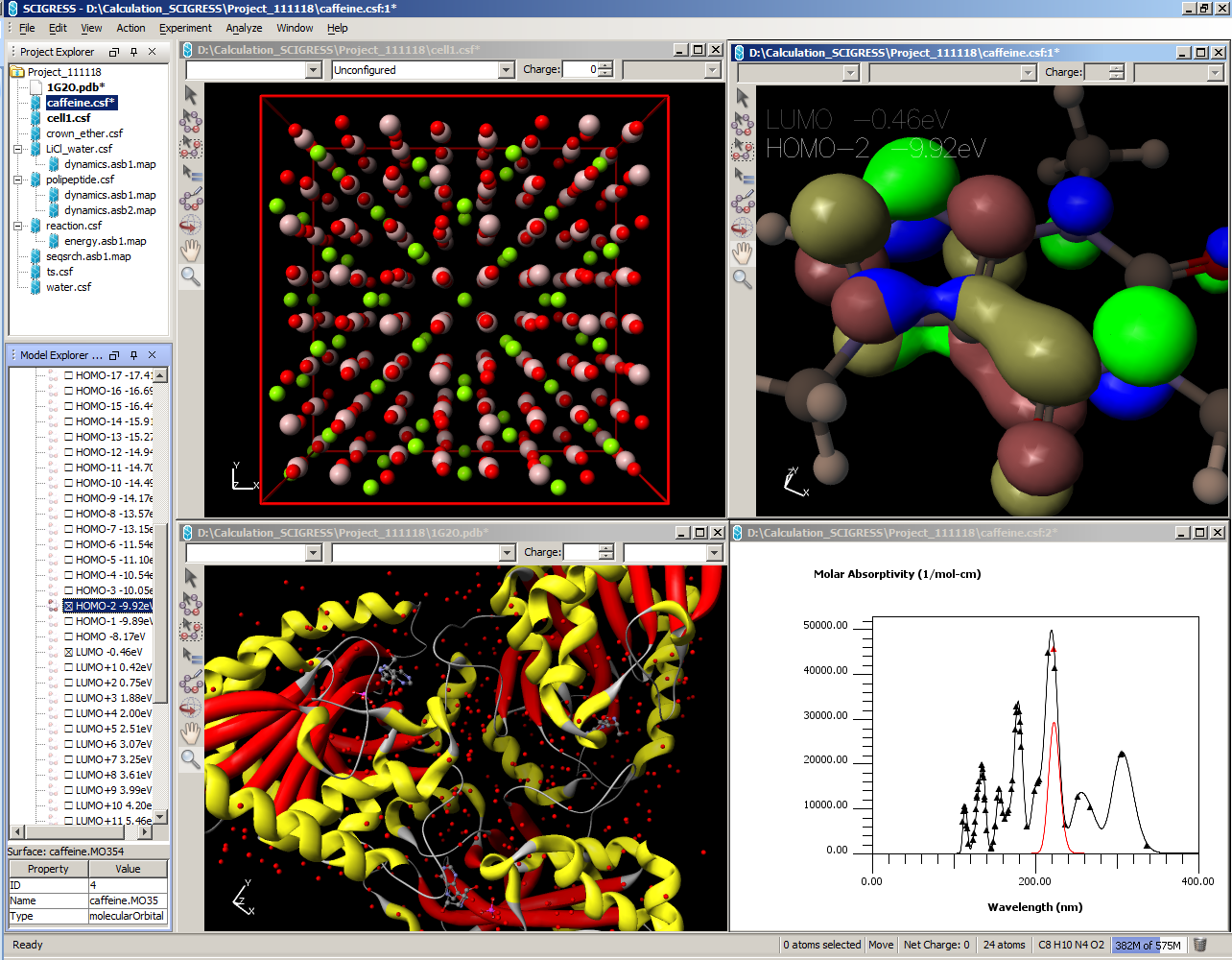
- Fully automated process of job submission and data retrieval and analysis:
- Local and remote calculations run at a press of a button.
- Multi-step simulations.
- Chemical Spreadsheet for batch jobs and fully automated QSAR analysis.
- Project explorer for tracking relations between input and output files.
- Graphical representation of computed properties:
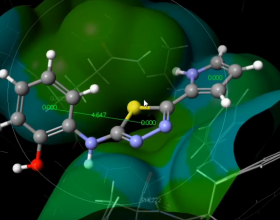
- Molecular editor/visualizer with highly customizable rendering options.
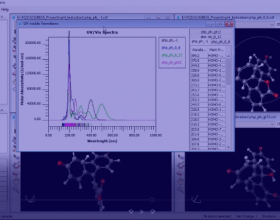
- Interactive spectra.
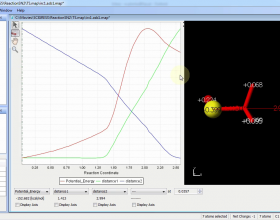
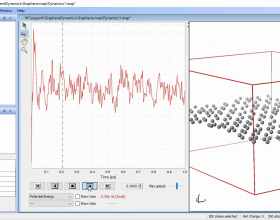
- Interactive potential energy graphs.
- Orbital and other electronic structural features visualization.
- Stereoscopic 3D visualization on compatible hardware.
- Set of easily customizable predefined procedures for common calculations.
- Seamless interface to external compute engines, both free and commercial:
- Amsterdam Modeling Suite.
- AutoDock (Tools and Vina).
- CONFLEX.
- GAMESS.
- Gaussian.
- LAMMPS.
- MOPAC.
- Quantum ESPRESSO.