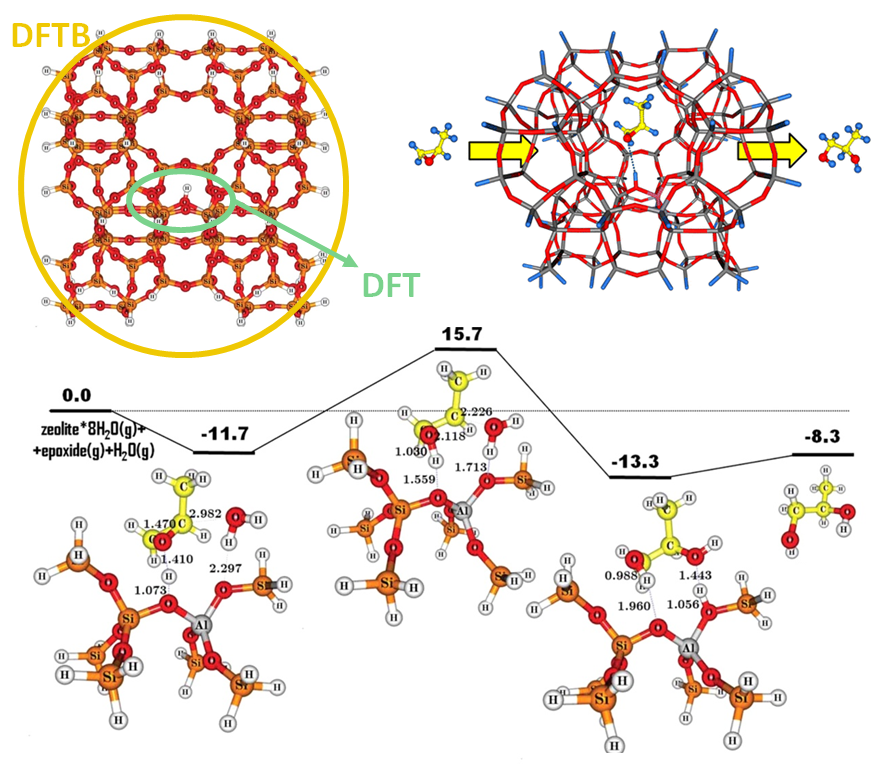
- Inorganic chemistry.
- Organic electronics.
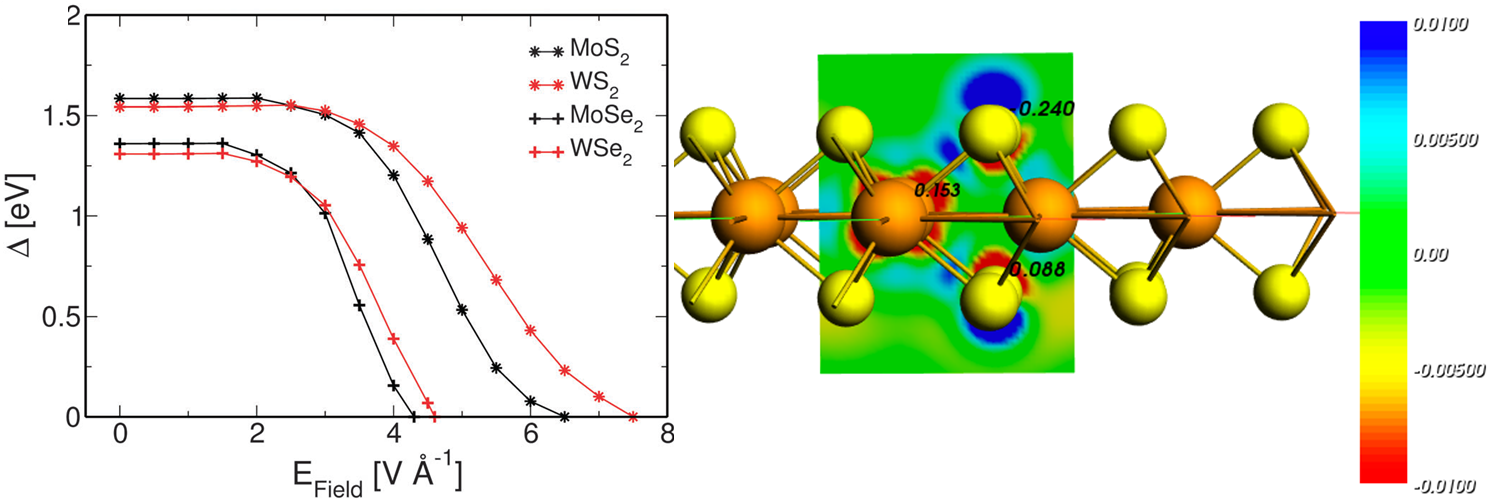
- Materials science.
- Nanoscience.
- Pharma and life sciences.
- Oil and gas.
- Spectroscopy.
- Heavy elements.
- Chemical bonding analysis.
- Batteries and photovoltaics.
- Catalysis.


Wkrótce się z Tobą skontaktujemy.

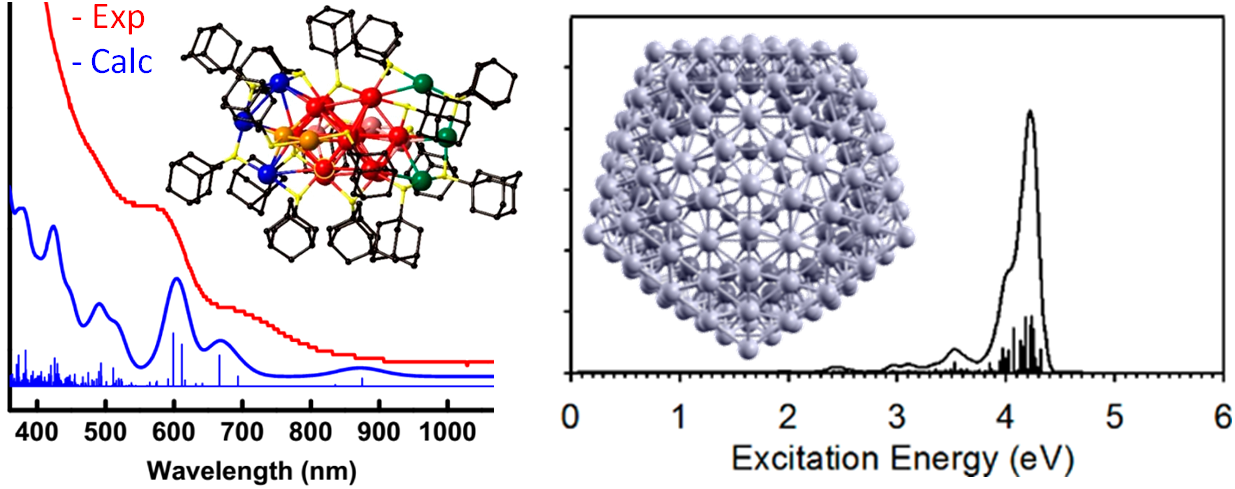
Relativistic TDDFT calculations on Au24(SAdm)16 and on icosahedral Ag2544+ monatomic nanoshell.
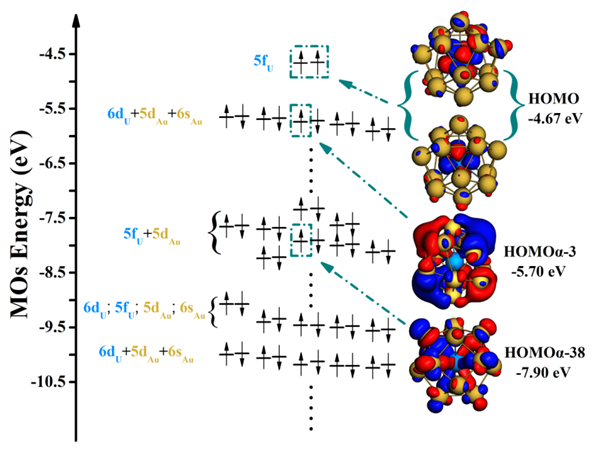
Electronic energy level diagram of ground state U@Au14.
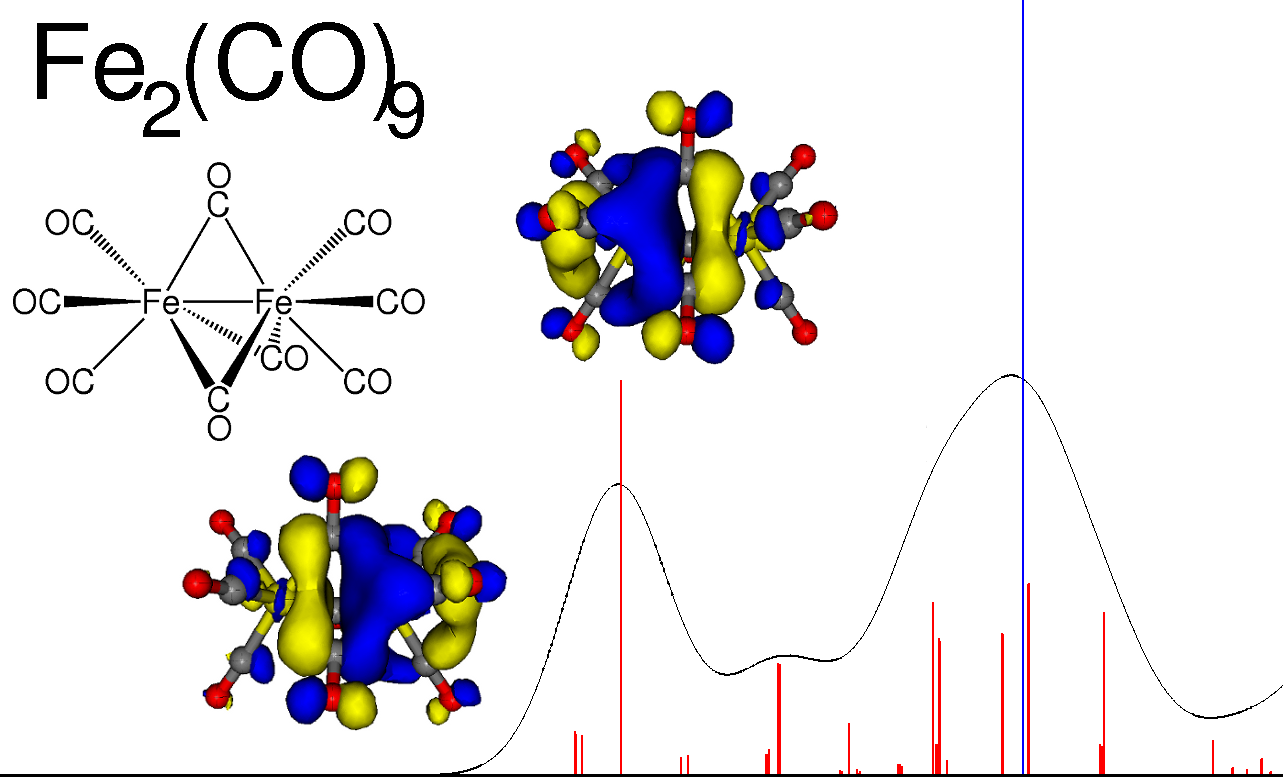
Calculated X-ray absorption spectrum for Fe2(CO)9 (absorption peaks in red, ionization blue).
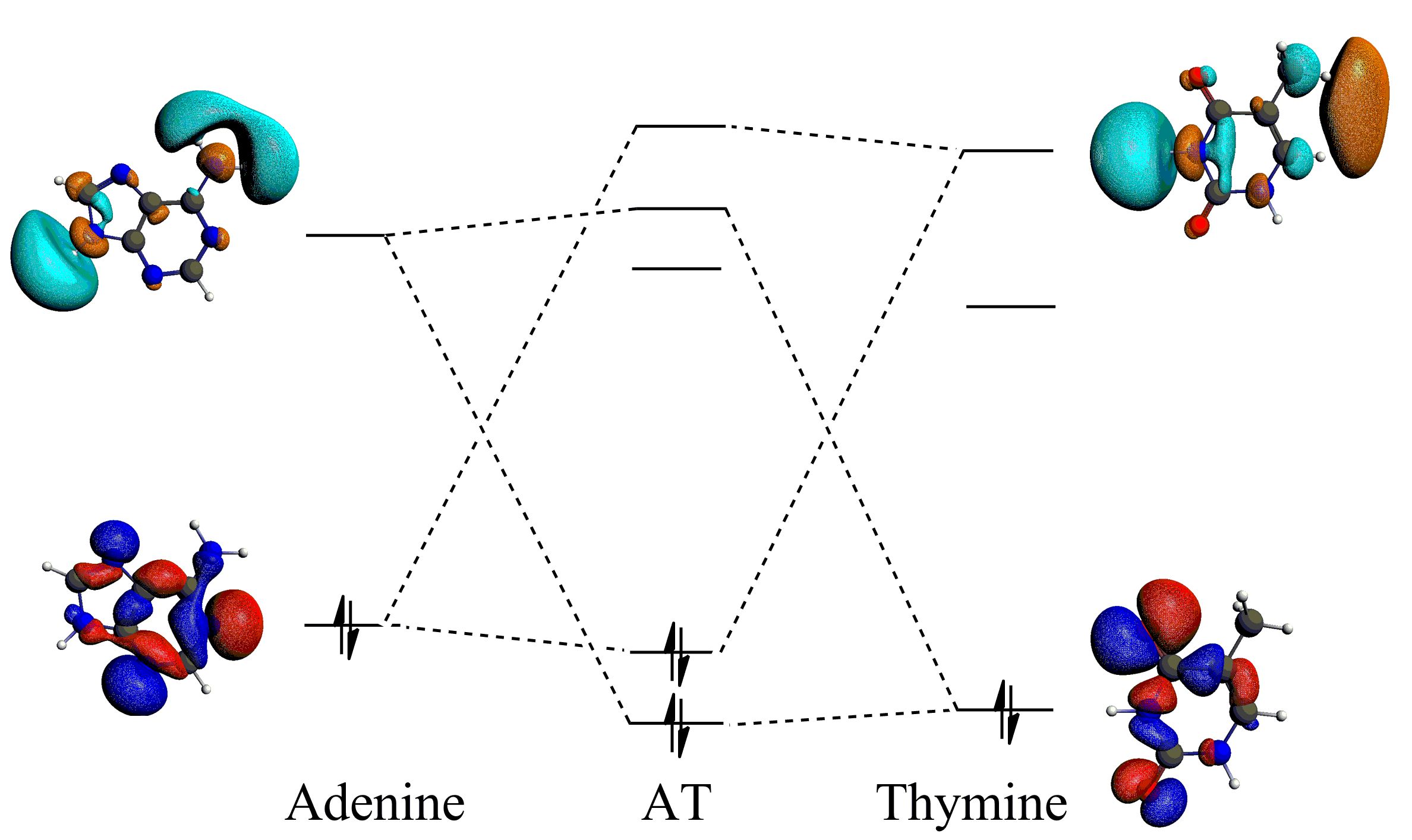
Donor-accpetor interactions for the hydrogen bonds in the AT base pair, enhanced by the sp2 hybridization.
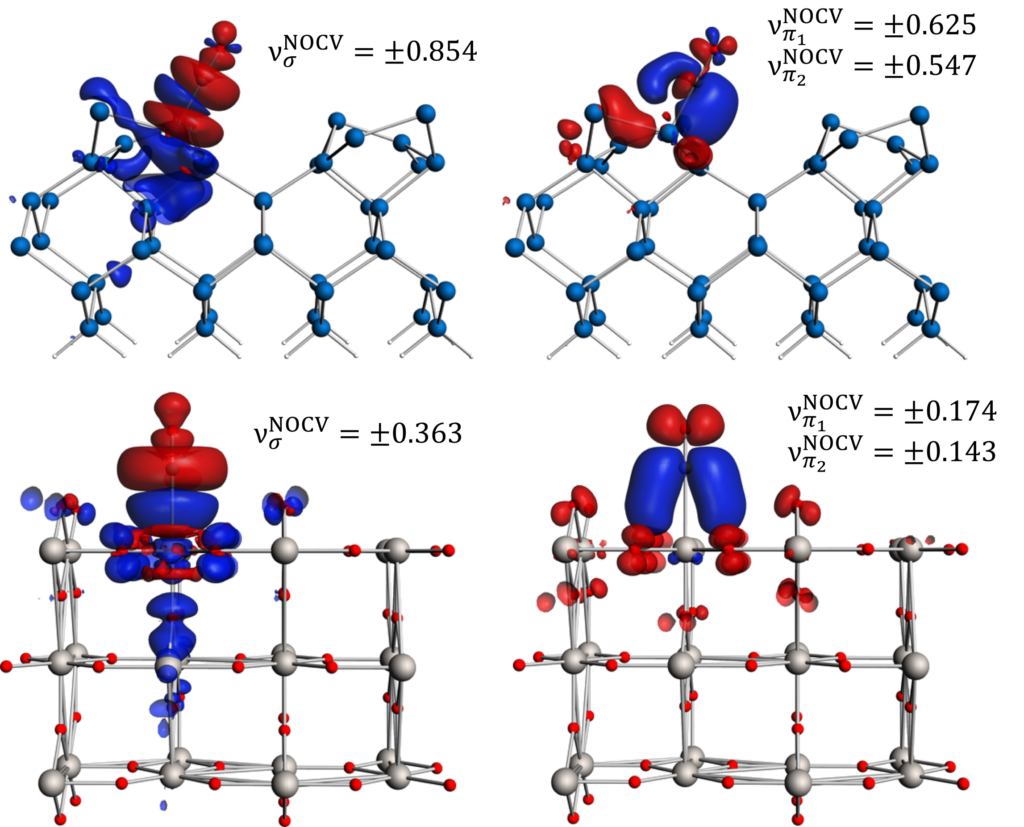
pEDA-NOCV: σ-donation from and π-back-donation to CO, adsorbed on Si (top) and TiO2 (bottom).
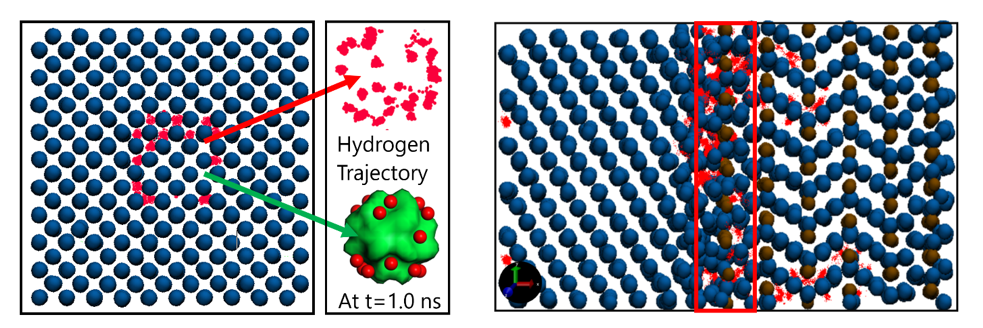
ReaxFF MD simulations for hydrogen trapping at the nanovoids and at the ferrite-cementite interfaces.
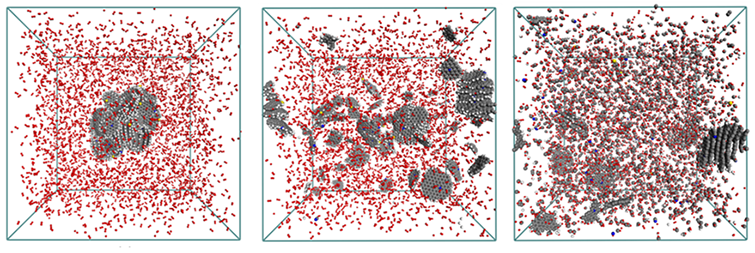
ReaxFF MD simulation of a combusting char structure surrounded by 14,000 O2 at 3000K (0, 75, and 250ps).
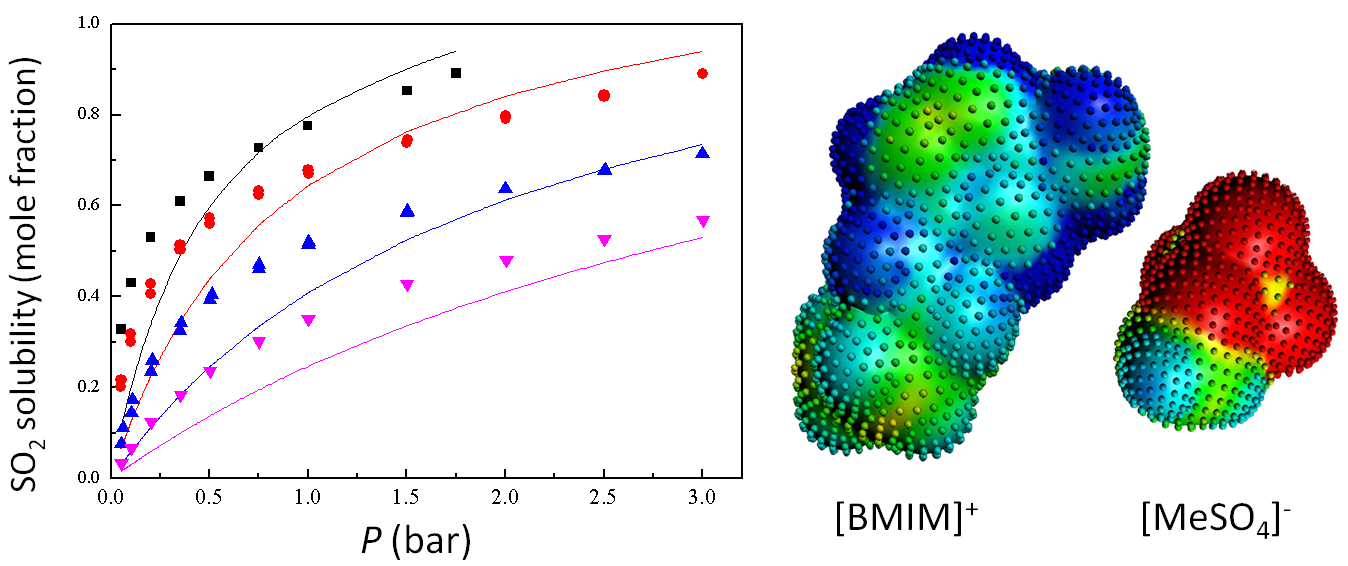
Prediction of SO2 solubilities in [BMIM]+[MeSO4]−, compared with experiments at different temperatures.
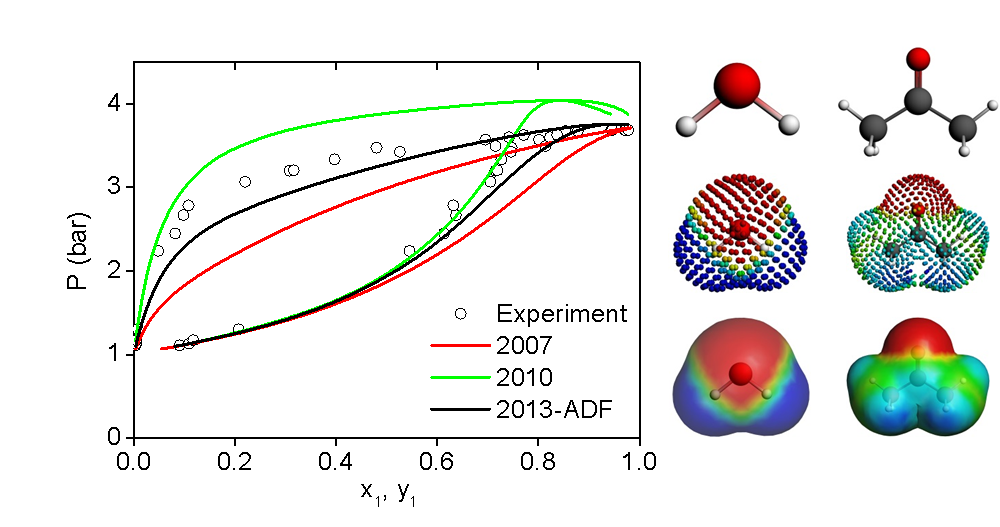
COSMO-SAC models and experimental vapor pressures for the acetone-water system.
Relativistic TDDFT calculations on Au24(SAdm)16 and on icosahedral Ag2544+ monatomic nanoshell.
Electronic energy level diagram of ground state U@Au14.
Calculated X-ray absorption spectrum for Fe2(CO)9 (absorption peaks in red, ionization blue).
Donor-accpetor interactions for the hydrogen bonds in the AT base pair, enhanced by the sp2 hybridization.
pEDA-NOCV: σ-donation from and π-back-donation to CO, adsorbed on Si (top) and TiO2 (bottom).
ReaxFF MD simulations for hydrogen trapping at the nanovoids and at the ferrite-cementite interfaces.
ReaxFF MD simulation of a combusting char structure surrounded by 14,000 O2 at 3000K (0, 75, and 250ps).
Prediction of SO2 solubilities in [BMIM]+[MeSO4]−, compared with experiments at different temperatures.
COSMO-SAC models and experimental vapor pressures for the acetone-water system.